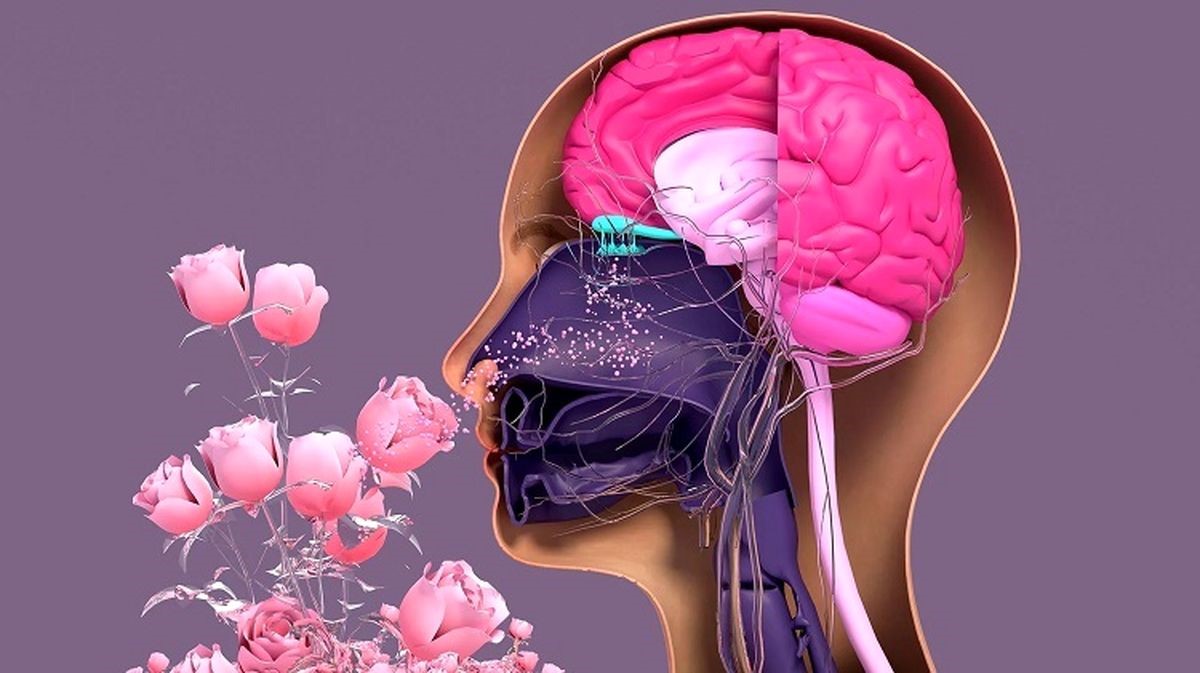
سندروم کالمن (Kallmann Syndrome) اختلال در حس بویایی را با یک اختلال هورمونی ترکیب میکند و بلوغ را به تأخیر میاندازد یا از آن جلوگیری میکند. این اختلال هورمونی که یک عارضه نادر ژنتیکی محسوب میشود، به دلیل توسعه نیافتن نورونها یا اعصاب خاصی در مغز به وجود میآید که به هیپوتالاموس سیگنال میدهند. بدون این نورونها، هیپوتالاموس نمیتواند تولید و ترشح هورمونهای خاص توسط غده هیپوفیز را بهدرستی تحریک کند.
برای درخواست هرگونه مشاوره، انجام آزمایش هورمونی و آزمایش ژنتیک و ثبت درخواست تلفنی نمونهگیری (در آزمایشگاه، منزل، محل کار)
با تلفن ۱۶۵۰ برای استان تهران و تلفن ۸۸۷۰۹۹۷۰-۰۲۱ برای دیگر استانها
با آزمایشگاه پاتوبیولوژی و ژنتیک پزشکی تهران لب تماس بگیرید.
آشنایی با Kallmann Syndrome
در رشد طبیعی، هیپوتالاموس هورمون آزادکننده گنادوتروپین (GnRH) را در دوران بلوغ ترشح میکند. این انفجارهای GnRH غده هیپوفیز را تحریک میکند تا هورمونهایی تولید کند که به نوبه خود باعث آزاد شدن هورمونهای جنسی مردانه و زنانه توسط غدد جنسی (بیضهها و تخمدانها) و رشد سلولهای اسپرم و تخمک میشود.
در سندروم کالمن، هیپوتالاموس نمیتواند هورمونهای GnRH را در رحم، در دوران نوزادی و در دوران بلوغ ترشح کند. احتمال بروز این بیماری، ۱ در ۴۸۰۰۰ نفر است. رشد اولیه هیپوتالاموس و حس بویایی در جنین انسان هر دو در سندروم کالمن تحت تأثیر قرار میگیرند.
بخشی از هیپوتالاموس که GnRH تولید میکند، در اصل ابتدا بهعنوان بخشی از بینی جنین شکل میگیرد و در مراحل اولیه رشد جنینی، تغییر مکان داده و به بقیه غده هیپوتالاموس میپیوندد. در Kallmann Syndrome، این بخش از هیپوتالاموس و نورونهای تشخیص بو (بویایی) در مغز بهطور کامل رشد نمیکنند.
زمانی که فردی دارای کمبود هورمون مشخصه این سندروم باشد؛ اما حس بویایی طبیعی داشته باشد، این وضعیت بهعنوان هیپوگنادیسم هیپوگنادوتروپیک ایدیوپاتیک نرموزمی(nIHH) شناخته میشود.
نامهای دیگر این اختلال عبارتاند از:
- هیپوگنادیسم آنوسمیک
- هیپوگنادیسم هیپوگنادوتروپیک ایدیوپاتیک آنوسمیک
- هیپوگنادیسم همراه با آنوسمی
- هیپوگنادیسم هیپوگنادوتروپ و آنوسمی
- سندروم هیپوگنادیسم-آنوسمیهیپوگنادوتروپیک
علل سندروم کالمن
Kallmann Syndrome و nIHH هر دو جزو اختلالهای ژنتیکی هستند و در اثر جهش در هر یک از چندین ژن مختلف ایجاد میشوند. جهشهای ژنی مختلف که منجر به Kallmann Syndrome و nIHH میشوند، الگوهای وراثتی متفاوتی دارند. برخی از این جهشها شناسایی شده و الگوهای وراثتی آنها ترسیم شده است.
برخی از آنها باید از هر دو والد به ارث برده شوند (ارث اتوزومال مغلوب). در این نوع، هر دو والدین ممکن است ناقل باشند؛ به این معنی که هیچ علامتی از بیماری ندارند. برخی دیگر را میتوان از مادر یا پدر به ارث برد (ارث غالب اتوزومی).
در انواع دیگر، مادر ناقل بوده و پدر دارای شرایط (ارث مرتبط با X) است. در این نوع از این سندروم، این بیماری از مادر به پسر منتقل میشود؛ اما از پدر به پسر منتقل نخواهد شد. هر یک از والدین میتوانند این شرایط را بهعنوان ناقل، به فرزند دختر منتقل کنند.
محققان اخیراً چهارمین الگوی توارثی را شناسایی کردهاند که در آن جهشهای بیش از یک ژن ممکن است با هم ترکیب شده و باعث ایجاد این بیماری شوند (ارث الیگوژنیک). محققان همچنان در حال کار برای شناسایی تمام جهشهای ژنتیکی مرتبط با Kallmann Syndrome و nIHH هستند.
در حالت کلی، ۲۵ ژن در بروز این اختلال دخیل هستند. جهش ژنتیکی در شش ژن زیر، بیشترین اثرگذاری را در بروز این بیماری خواهند داشت:
- ANOS1
- CHD7
- FGF8
- FGFR1
- PROK2
- PROKR2
علائم و نشانههای سندروم کالمن
علامت اصلی این سندروم، تأخیر در بلوغ یا بلوغ ناقص است. در این اختلال،حس بویایی نوزاد بهصورت کامل یا جزئی مختل میشود. وضعیتی که از بدو تولد وجود دارد؛ اما اغلب تا زمانی که در طول تشخیص علت بلوغ تأخیری در مورد آن سوال نشود، تشخیص داده نخواهد شد.
این اختلال غالباً در سن ۱۴ تا ۱۶ سالگی قابل تشخیص است. از دیگر نشانههای وجود این عارضه در نوزاد و کودک، میتوان به موارد زیر اشاره کرد:
- بیضههای افتاده یا تا حدی نزولشده
- اندازه آلت تناسلی کوچک
- نقصهای صورت، مانند شکاف لب یا کام
- کوتاهی انگشتان دست یا پا به خصوص انگشت چهارم
- رشد تنها یک کلیه
- از دست دادن شنوایی
- کور رنگی
- حرکات غیرطبیعی چشم
- رشد غیرطبیعی دندانها
- حرکات دست آینهای (سینکینز دو دستی)، که در آن حرکات یک دست توسط دست دیگر تقلید میشود.
- عدم تشکیل کلیه
- از دست دادن شنوایی
- عمر کوتاهتر
آزمایش و تشخیص سندروم کالمن
پزشک معمولاً با یک معاینه فیزیکی و با سؤالاتی روند تشخیص را آغاز میکند که ممکن است متوجه آن شده باشید. علائم بلوغ تأخیری یا بلوغ ناقص و اختلال در حس بویایی در صدر این پرسشها و معاینههای بالینی قرار دارند.
از آنجایی که این اختلال یک بیماری ارثی است، پزشک ممکن است در مورد بستگانی که بلوغ تأخیری یا مشکلات باروری را تجربه کردهاند نیز سؤال کند. اگر علائم بیمار و مشاهدات پزشک، احتمال سندروم کالمن یا nIHH را نشان دهد، آزمایشهای اضافی و تکمیلی برای مشاوره ژنتیک بهتر از بیمار گرفته خواهند شد.
این آزمایشها که میتوانند در آزمایشگاه و بیمارستان انجام شوند، شامل موارد زیر هستند:
- آزمایش هورمونی: بهطور خاص برای بررسی سطح هورمون در وریدهای محیطی که از غده هیپوفیز منشأ میگیرند.
- تصویربرداری رزونانس مغناطیسی (MRI) از هیپوتالاموس: غده هیپوفیز و بینی برای بررسی ناهنجاریهای آناتومیکی مورد تصویربرداری قرار میگیرند.
- آزمایش ژنتیک مولکولی: برای جستجوی جهشهای ژنی خاص

درمان سندروم کالمن
Kallmann Syndrome و nIHH با درمان جایگزینی هورمونی، به کمک داروها و دوزهای خاص و متناسب با نیاز بیمار آغاز میشوند. این درمان ابتدا بر القای بلوغ و حفظ سطح طبیعی هورمون تمرکز دارد. سپس ممکن است روند درمان بر روی القای باروری متمرکز شود.
همچنین ممکن است بیمار برای بازیابی سلامت استخوانها، به دارو درمانی نیاز داشته باشد؛ زیرا عدم وجود هورمونهای کلیدی که منجر به تأخیر در بلوغ میشود، میتواند منجر به تضعیف تراکم و استحکام استخوانها نیز شده باشد.
در درازمدت، درمان جایگزینی هورمون برای مردان، ممکن است بهطور دورهای کاهش یافته یا متوقف شود. این توقف و کاهش برای تشخیص و رصد این موضوع است که آیا بدن شرایط را معکوس کرده و سطوح طبیعی هورمونها را تولید میکند یا خیر.
هنگامی که دارو درمانی برای تکمیل تولید هورمون مورد نیاز باشد، آزمایشهای پیگیری دورهای برای اطمینان از ادامه اثربخشی درمان مورد نیاز خواهد بود. سطح دوز و ترکیب داروها ممکن است در طول زمان، نیاز به تنظیم داشته باشد.
همچنین، درمان همزمان مشکلات جسمی و ذهنی فرد و ارائه خدمات حمایتی و آموزشی به خانوادهها نیز بسیار مهم است. گفتنی است که هر فرد مبتلاء به Kallmann Syndrome دارای نیازها و ویژگیهای منحصربهفرد خود است. بنابراین، درمان و مراقبت باید بر اساس نیازهای بیمار شخصیسازی شود.
این بیماری نیازمند مراقبت مداوم و توجه ویژه است و مدیریت آن باید توسط یک تیم متخصص از پزشکان، متخصصین تغذیه، روانشناسان و سایر متخصصین بهداشتی انجام شود.
برای درخواست هرگونه مشاوره، انجام آزمایش و ثبت درخواست تلفنی نمونهگیری (در آزمایشگاه، منزل، محل کار)
با تلفن ۱۶۵۰ برای استان تهران و تلفن ۸۸۷۰۹۹۷۰-۰۲۱ برای دیگر استانها
با آزمایشگاه پاتوبیولوژی و ژنتیک پزشکی تهران لب تماس بگیرید.
آنچه در مقاله سندروم کالمن آموختیم
سندروم کالمن یک بیماری ژنتیکی نادر است که بهطور غالب از طریق مادر به فرزندان انتقال مییابد. این بیماری باعث ایجاد عوارض جسمی و ذهنی جدی میشود. بیماران مبتلاء به Kallmann Syndrome معمولاً دارای مشکلات قلبی، تأخر در رشد جسمی و ذهنی، نقصهای فیزیکی، مشکلات دستگاه ادراری و مشکلات بویایی هستند. این بیماری با تحلیل ژنتیکی و مطالعه ویژگیهای جسمی فرد تشخیص داده میشود. درمان این بیماری بیشتر برای کنترل علائم و تسریع در رشد و توسعه فیزیکی و ذهنی فرد است.
- منابع: