بیماری فنیل کتونوری (PKU) یک اختلال ژنتیکی نادر و مزمن است که به دلیل جهش در ژن PAH ایجاد میشود. ژن PAH دستورالعمل تولید آنزیم فنیل آلانین هیدروکسیلاز را در بدن فراهم میکند. این آنزیم به تجزیه آمینواسید فنیل آلانین کمک مینماید. اما در افراد مبتلاء به فنیل کتونوری، تغییرات ژنتیکی باعث کاهش یا فقدان عملکرد این آنزیم میشود، که در نتیجه آن سطح فنیل آلانین در خون و بافتهای بدن به میزان غیرطبیعی بالا میرود.
تجمع فنیل آلانین میتواند به سیستم عصبی و مغز آسیب برساند و اگر به موقع تشخیص و مدیریت نشود، منجر به ناتوانیهای ذهنی، مشکلات رفتاری و سایر عوارض جدی میگردد.
روشهای تشخیص ژن PAH و آزمایشهای تشخیصی بیماری فنیل کتونوری ژن PAH نقشی کلیدی در تشخیص زودهنگام و مدیریت این بیماری دارند. با شناسایی جهشهای ژن PAH، میتوان برای بیماران درمانهای مناسبتری را ارائه و رژیمهای غذایی کنترل شده را اعمال کرد.
در این مقاله توضیحاتی در مورد بیماری فنیل کتونوری، انواع بیماری PKU، علت بروز این بیماری، جهشهای ژن PAH، روشهای تشخیص ژن PAH و آزمایشهای تشخیصی بیماری فنیل کتونوری ژن PAH و مدیریت و درمان بیماری فنیل کتونوری توضیحاتی بیان میشود.
بیماری فنیل کتونوری چه نوع بیماری است؟
فنیل کتونوری (PKU یا Phenylketonuria) یک بیماری متابولیکی ژنتیکی (ناشی از جهش ژن PAH) است که باعث افزایش غیرطبیعی سطح فنیل آلانین در بدن میشود. فنیل آلانین یک آمینواسید میباشد که در مسیرهای بیوشیمیایی طبیعی بدن وجود دارد و از مواد تشکیلدهنده پروتئینها است. این ماده در بسیاری از غذاهای پروتئینی و همچنین در شیرینکننده مصنوعی آسپارتام یافت میشود.
در شرایط عادی، بدن فنیل آلانین را به طور طبیعی پردازش میکند، اما در افراد مبتلاء به فنیل کتونوری یا PKU، سطح فنیلآلانین در بدن بالا میرود. اگر این بیماری مدیریت نشود، تجمع فنیل آلانین میتواند منجر به مشکلاتی (مانند ناتوانیهای ذهنی) گردد.
در آمریکا، تخمین زده میشود که این بیماری از هر ۱۰,۰۰۰ تا ۱۵,۰۰۰ نوزاد، یک نفر را هر ساله تحت تأثیر قرار میدهد.
انواع بیماری فنیل کتونوری
فنیل کتونوری براساس شدت بیماری سه نوع میباشد: فنیل کتونوری کلاسیک، فنیل کتونوری متوسط یا خفیف و هایپر فنیل آلانینمی خفیف. در ادامه راجع به هر کدام توضیحاتی ارائه میشود:
- فنیل کتونوری کلاسیک (شدیدترین نوع): این نوع فنیل کتونوری، شدیدترین شکل PKU است که در آن آنزیم مورد نیاز برای تجزیه فنیل آلانین وجود ندارد یا به شدت کاهش مییابد. این وضعیت منجر به سطوح بالای فنیل آلانین میشود که میتواند آسیبهای جدی به مغز وارد کند.
- فنیل کتونوری متوسط یا خفیف: در این نوع فنیل کتونوری ، آنزیم فنیل آلانین هیدروکسیلاز (PAH) هنوز هم عملکردی دارد، بنابراین سطح فنیل آلانین نسبتاً پایین است و خطر آسیب مغزی کمتر میباشد.
- هایپر فتیل آلانینمی خفیف (خفیفنوین نوع): در بین انواع بیماری فتیل کتونوری، هایپر فتیل آلانینمی خفیف کمترین شدت را دارد.
علت بروز بیماری PKU چیست؟
بیماری فنیل کتونوری (PKU) به دلیل بروز جهش و تغییرات در ژن PAH (فنیل آلانین هیدروکسیلاز) ایجاد میشود. این ژن مسئول تولید آنزیمی به نام فنیل آلانین هیدروکسیلاز است که فنیلآلانین را به ترکیبات مهم دیگری در بدن تبدیل میکند. اگر کودک دو نسخه از ژن جهشیافته (یک نسخه از هر والد) را به ارث ببرد، به این بیماری مبتلاء خواهد شد.
بروز جهش در در هر دو نسخه از ژن PAH موجب تولید نسخههای تغییریافتهای از فنیل آلانین هیدروکسیلاز میشود که قادر به پردازش فنیل آلانین بهطور موثر نیست. این وضعیت منجر به تجمع فنیل آلانین در خون و بافتهای دیگر و رسیدن به سطح سمی میشود. از آنجا که سلولهای عصبی در مغز به میزان فنیل آلانین بسیار حساس هستند، تجمع بیش از حد این ماده میتواند به آسیب مغزی منجر شود.
در ادامه، در خصوص جهشهای ژن PAH توضیحاتی ارائه میشود.
جهشهای ژن PAH
برای ژن PAH تاکنون بیش از ۵۰۰ نوع جهش شناسایی شده است و با استفاده از روش توالییابی دوطرفه، میتوان ۹۹% از جهشهای مناطق کدکننده (اگزونهای ۱ تا ۱۳) را شناسایی کرد. انواع اصلی جهشهای ژن PAH شامل موارد زیر است:
- جهشهای جایگزینی (Missense Mutations): جهشهای جایگزینی در کدونهای خاصی از ژن رخ میدهند و موجب تغییر یک آمینواسید به آمینواسید دیگر میشوند، که میتواند باعث اختلال در ساختار و عملکرد آنزیم فنیل آلانین هیدروکسیلاز گردد.
- جهشهای بیمعنی (Nonsense Mutations): این جهشها باعث میشوند که کدون توقف زودهنگام ایجاد شود و تولید آنزیم ناقص و کوتاه گردد. این نوع جهشها معمولاً منجر به عملکرد بسیار کم یا عدم عملکرد آنزیم میشوند.
- حذف یا اضافه شدن (Insertions or Deletions): این نوع جهشها شامل حذف یا اضافه شدن نوکلئوتیدها هستند و باعث تغییر در ساختار کلی ژن و ایجاد تغییرات گسترده در آنزیم تولیدشده میشوند.
- جهشهای چارچوبی (Frameshift Mutations): جهشهای چارچوبی باعث تغییر در چارچوب خوانش ژن میشوند و میتوانند منجر به تولید آنزیمهای غیرفعال شوند.
در مورد روشهای تشخیص ژن PAH و آزمایشهای تشخیصی بیماری فنیل کتونوری ژن PAH، در ادامه توضیحاتی بیان خواهد شد. انتخاب بهترین آزمایشگاه تهران، برای انجام آزمایشات ژنتیکی و خون به تشخیص درست بیماری و انجام اقدامات درمانی مناسب کمک میکند.

علائم بیماری فنیل کتونوری
بیماری فنیل کتونوری در بیشتر موارد، به دلیل انجام غربالگری نوزادان پس از تولد، زود تشخیص و مدیریت میگردد و به همین دلیل علائم مشهود آن بسیار نادر است. علائم بیماری PKU معمولاً در مواردی که این بیماری تشخیص و مدیریت نشود، مشاهده میگردد. برخی از علائم این بیماری عبارت است از:
- اگزما
- تغییر رنگ پوست یا مو (روشنتر از دیگر اعضای خانواده)
- اندازه کوچک سر (میکروسفالی)
- بوی کپک مانند در نفس، پوست یا ادرار
علائم شدید فنیل کتونوری درمان نشده هم شامل موارد زیر است:
- مشکلات رفتاری
- تأخیر در رشد
- ناتوانیهای ذهنی
- تشنج (در مواقع نادر)
نکته: کودکان و بزرگسالانی که به هایپر فنیل آلانینمی خفیف مبتلاء هستند، در صورت عدم مدیریت، خطر کمتری برای ابتلاء به ناتوانیهای ذهنی دارند.
روشهای تشخیص ژن PAH و فنیل کتونوری
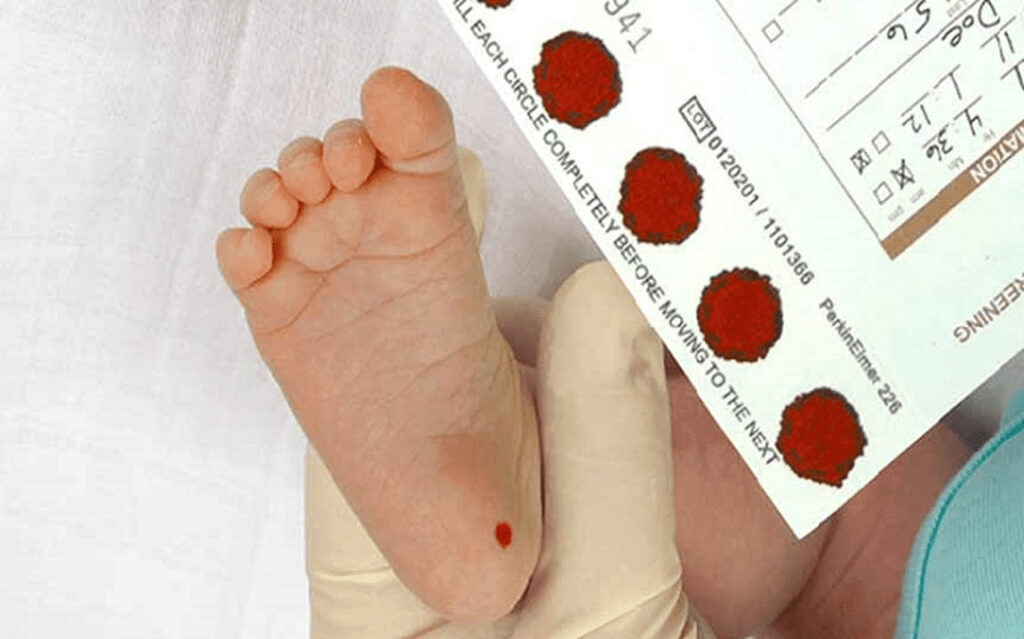
برای تشخیص بیماری فنیل کتونوری نیاز به انجام آزمایشهای تشخیصی بیماری فنیل کتونوری ژن PAH است. معمولاً بلافاصله پس از تولد و به عنوان بخشی از غربالگریهای معمول نوزادان، یک آزمایش خون انجام میشود. این آزمایش سطح فنیلآلانین خون را اندازهگیری میکند.
اگر نتایج این آزمایش نشان دهد که سطح فنیلآلانین در نمونه خون نوزاد بالا است، پزشک برای تأیید تشخیص و تعیین نوع PKU، آزمایشهای تکمیلی دیگری انجام میدهد که معمولاً شامل آزمایشهای خون یا ادرار اضافی میباشد. از آنجایی که فنیل کتونوری یک بیماری ژنتیکی است، آزمایش ژنتیکی نیز میتواند نوع جهشی را که منجر به بروز علائم میشود، شناسایی کند.
نکته: اگرچه تشخیص بیماری PKU، در بیشتر مواقع بلافاصله پس از تولد انجام میشود، با این حال امکان تشخیص بیماری فنیل کتونوری در هر سنی وجود دارد.
مدیریت و درمان بیماری فنیل کتونوری
فنیل کتونوری، یک بیماری مادم العمر است و درمان قطعی ندارد، اما میتوان آن را با رژیم غذایی کنترل کرد. این بیماری، که به دلیل رخ دادن جهش در ژن PAH بروز مییابد، را میتوان با اتخاذ رژیم غذایی مناسب و مصرف داروها و مکملها مدیریت نمود. در ادامه، در خصوص رژیم غذایی مناسب برای فنیل کتونوری و داروهای این بیماری توضیحاتی ارائه میشود.
رژیم غذایی مناسب برای بیماری فنیل کتونوری
افراد مبتلاء به این بیماری باید از رژیم غذایی خاصی استفاده کنند که در کنار غنی بودن از مواد مغذی، فنیل آلانین کمی داشته باشد. افراد مبتلاء به PKU باید از مصرف خوراکیها و غذاهای غنی از پروتئین که سرشار از فنیل آلانین هستند، پرهیز کنند (مگر اینکه تحت درمان با Pegvaliase باشند). از جمله خوراکیها و موادغذاییهای مضر برای مبتلایان به فنیل کتونوری میتوان به موارد زیر اشاره کرد:
- شیر
- تخممرغ
- پنیر
- آجیل
- ماهی
- مرغ
- گوشت گاو
- لوبیا
- شیرینکنندههای مصنوعی (آسپارتام)
نکته: برای جبران کمبودهای بدن، مصرف ویتامینها و مواد معدنی توصیه میشود.
داروهای فنیل کتونوری
داروی ساپروپترین دی هیدروکلراید (کووان یا Kuvan) و داروی Pegvaliase (Palynziq)، از جمله داروهای موثر بر فنیل کتونوری هستند. ساپروپترین به تجزیه فنیل آلانین در بدن کمک میکند و برای برخی از بیماران میتواند مؤثر باشد.
Pegvaliase هم برای بیماران PKU این امکان را فراهم میکند که بدون نیاز به رژیم غذایی محدود، مکملها یا Kuvan، بتوانند به رژیم غذایی عادی روی بیاورند. این دارو جایگزین آنزیم تجزیهکننده فنیل آلانین میشود که در افراد مبتلاء به فنیل کتونوری به درستی عمل نمیکند.
با استفاده از روشهای تشخیص ژن PAH و آزمایشهای تشخیصی بیماری فنیل کتونوری ژن PAH، میتوان به طور دقیق این بیماری را شناسایی و کنترل کرد. انتخاب بهترین آزمایشگاه در تهران برای انجام آزمایشات ژنتیکی و خون، به جهت برخورداری از کادر حرفهای و تجهیزات آزمایشگاهی پیشرفته، سبب کسب نتایج آزمایشگاهی دقیق و اطلاع درست از وضعیت سلامت میشود.
برای درخواست هرگونه مشاوره ژنتیکی تلفنی و نمونهگیری در آزمایشگاه جهت انجام آزمایش تشخیص فنیل کتونوری، با تلفن ۱۶۵۰ آزمایشگاه پاتوبیولوژی و ژنتیک پزشکی تهران لب تماس بگیرید.
آنچه از مقاله «بیماری فنیل کتونوری چیست؟ | بررسی جهشهای ژن PAH» آموختیم
همان طور که بیان شد، بیماری فنیل کتونوری (PKU) یک اختلال متابولیکی ژنتیکی ناشی از جهشهای ژن PAH (فنیل آلانین هیدروکسیلاز) میباشد. این ژن نقشی حیاتی در تولید آنزیم تجزیهکننده آمینواسید فنیل آلانین دارد، که در صورت بروز اختلال، سطح فنیل آلانین در خون به شدت افزایش مییابد و این موضوع به تدریج سبب آسیب به سیستم عصبی میشود.
با توجه به پیامدهای جدی تجمع فنیل آلانین بر مغز و تواناییهای شناختی، روشهای دقیق تشخیص ژن PAH و آزمایشهای تشخیصی بیماری فنیل کتونوری ژن PAH، برای شناسایی و مدیریت این بیماری اهمیت فراوانی دارند.
امروزه آزمایشهای ژنتیکی پیشرفته و غربالگریهای نوزادان پس از تولد امکان تشخیص زودهنگام و در نتیجه کنترل بهتر بیماری را فراهم کردهاند. این بررسیها، جهشهای ژنی را شناسایی کرده و درجه شدت بیماری را مشخص میکنند که در کنار درمانهایی همچون رژیمهای غذایی و داروهای نوین مانند ساپروپترین دی هیدروکلراید (کووان) و داروی Pegvaliase، به بیماران کمک میکند تا زندگی عادیتری داشته باشند.
انتخاب بهترین آزمایشگاه برای انجام آزمایش غربالگری نوزاد جهت تشخیص دقیق بیماری فنیل کتونوری، نقش موثری در بهبود کیفیت زندگی مبتلایان به PKU ایفاء میکند.
- منابع:
https://my.clevelandclinic.org/health/diseases/17816-phenylketonuria